


Mapping algorithms and softwares
This analysis was performed using R (ver. 3.1.0).FASTQ file
FASTQ file, is the result of a next-generation sequencing experiment. Every four lines, indicates a read. The first line gives the name of the read and some other information, including its length. The second line is the base pairs of the read. The fourth line contains information about the quality of each base pair. The third line links all the qualities to the first line.
FASTQ Format
A FASTQ file normally uses four lines per sequence.
- Line 1 begins with a ‘@’ character and is followed by a sequence identifier and an optional description (like a FASTA title line).
- Line 2 is the raw sequence letters.
- Line 3 begins with a ‘+’ character and is optionally followed by the same sequence identifier (and any description) again.
- Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence.
A FASTQ file containing a single sequence might look like this:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65Mapping Algorithms and Softwares
Mapping software is also called alignment software. One of the most popular short read aligners is Bowtie. Help can be found on Bowtie website or from linux command: bowtie –help.
Download sequencing data and extract the FASQ files
Download RNA sequencing reads from Short Read Archive (SRA) :
GSE52166, SRP032775, http://www.ncbi.nlm.nih.gov/Traces/sra/?run=SRR1177756.
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR117/SRR1177756/SRR1177756.sraWe use the software fastq-dump to extract the FASTQ files from the .sra file :
fastq-dump --split-file-3 SRR1177756.sra
# view generated files with size
ls -lh *.fastqThe option --split-file-3 is used for paired-end sequencing.Two FastQ files are generated (SRR1177756_1.fastq, SRR1177756_2.fastq), because data is a paired-end sequencing. Each read is paired with the same name in each file. Align RNA sequencing data using Tophat
TopHat software is used to align RNA sequencing data. It takes care of the fact that the reads might span an intron. The TopHat software actually uses Bowtie internally to do part of the mapping.
To use TopHat, you need a Bowtie index, which is a precompiled version of the genome, which is efficiently built so that you can map these short reads against it. Bowtie index can be downloaded for different species on TopHat website.
Download reference genome from tophat website:
wget ftp://igenome:G3nom3s4u@ussd-ftp.illumina.com/Homo_sapiens/Ensembl/GRCh37/Homo_sapiens_Ensembl_GRCh37.tar.gz
tar zxvf Homo_sapiens_Ensembl_GRCh37.tar.gzRunning TopHat:
Tophat needs the path to reference genome. This path might be different on your computer.
tophat2 -o SRR117756_out -p 10 genomes/Homo_sapiens/Ensembl/GRCh37/Sequence/Bowtie2Index SRR1177756_1.fastq SRR1177756_2.fastqThe 2 files indicate left and right paired-end reads.
In the call to tophat2, the option -o specifies the output directory, -p specifies the number of threads to use (this may affect run times and can vary depending on the resources available).Use bioinformatic cluster to align reads
A bioinformatic cluster is needed to process huge data generated by next generation sequencing. I use genotool plateform to run my alignement experiment. The aim of the following section is to show, how to submit a job to genotoul. You can skip this section
Submit job to genotoul
My working directory hierarchy:
- work
- akassambara
- seq
- genomes : contains reference genome
- SRR117756_out
- SRR1177756_1.fastq
- SRR1177756_2.fastq
- SRR1177756.sra
- tophat_SRR1177756.sh
Preparing my script (tophat_SRR1177756.sh) to run on genotool:
#!/bin/bash
export BOWTIE2_INDEXES=/work/akassambara/seq/genomes/Homo_sapiens/Ensembl/GRCh37/Sequence/Bowtie2Index
tophat2 -o SRR117756_out -p 10 genomes/Homo_sapiens/Ensembl/GRCh37/Sequence/Bowtie2Index/genome SRR1177756_1.fastq SRR1177756_2.fastqSubmit the job
qsub -q workq -l h_vmem=100G -l mem=100G tophat_SRR1177756.sh Tophat result files
TopHat generates a list of files which are located in SRR117756_out folder.
akassambara@genotoul ~/work/seq/SRR117756_out $ ls -lh
total 4,1G
-rw-r--r-- 1 akassambara U1040 2,2G 30 juil. 05:12 accepted_hits.bam
-rw-r--r-- 1 akassambara U1040 9,5M 30 juil. 04:57 deletions.bed
-rw-r--r-- 1 akassambara U1040 7,4M 30 juil. 04:57 insertions.bed
-rw-r--r-- 1 akassambara U1040 11M 30 juil. 04:57 junctions.bed
drwxr-xr-x 2 akassambara U1040 16K 30 juil. 05:12 logs
-rw-r--r-- 1 akassambara U1040 188 29 juil. 16:59 prep_reads.info
-rw-r--r-- 1 akassambara U1040 1,9G 30 juil. 05:24 unmapped.bamaccepted_hits.bam file
This is a compressed file. It contains the reads which could be successfully mapped to the genome. You can use the samtools view function to read this compressed file.
The following line code can be used to view the first 1000 lines. Use “return” to view more and type q to quit.
#Look at the first 1000 lines, and then use less.
#You have to type q to quit less command
samtools view accepted_hits.bam | head -1000 | lessSRR1177756.30816791 393 1 12006 0 101M * 0 0 GCAGGTGTCTGACTTCCAGCAACTGCTGGCCTGTGCCAGGGTGGAAGCTGAGCACTGGAGTGGAGTTTTCCTGTGGAGAGGAGCCATGCCTAGAGTGGGAT BBBBFThis gives each read with the name of the read on the left, and where it was possible to align the read to the genome. For example, the read SRR1177756.30816791 was aligned to chromosome 1 at position 12,006. We could see that this whole read aligned (101 matches).
Down below, we see that there are reads where the whole read didn't align. If we consider that we're looking at RNA sequencing, and that some of these reads come from RNA pieces, which span junction in the genome, it makes sense that there would be parts of these which wouldn't align to the genome. So instead of a match, we get an N, which indicates there was a gap: 10M385N91M = 10 matches then 385 nucleotide gaps then 91 matches. The following SAMtools calls can be used to process the BAM files.
samtools sort -n accepted_hits.bam accepted_hits_name_sorted
samtools sort -n unmapped.bam unmapped_name_sorted
samtools merge -n all_reads.bam accepted_hits_name_sorted unmapped_name_sortedThe -n option indicates that I want to sort these reads, not by genomic location but, by name of the read. Sorted reads are put in the accepted_hits_name_sorted file.
Sorted reads:
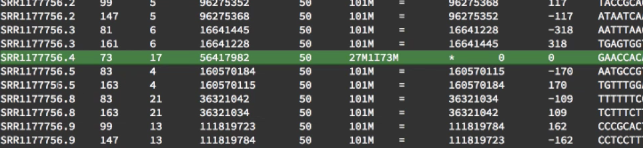
After sorting, you can see something interesting, which is that the fragment number 4 only has one read in this file. And also you can see that fragment number 1, and 6, and 7 are also not in this file. If we want to find those reads, we can look into the unmapped.bam.
If we want a single file with all of the alignments in it, we can use the samtools merge command. Licence
References
Show me some love with the like buttons below... Thank you and please don't forget to share and comment below!!
Montrez-moi un peu d'amour avec les like ci-dessous ... Merci et n'oubliez pas, s'il vous plaît, de partager et de commenter ci-dessous!
Recommended for You!





Recommended for you
This section contains best data science and self-development resources to help you on your path.
Coursera - Online Courses and Specialization
Data science
- Course: Machine Learning: Master the Fundamentals by Standford
- Specialization: Data Science by Johns Hopkins University
- Specialization: Python for Everybody by University of Michigan
- Courses: Build Skills for a Top Job in any Industry by Coursera
- Specialization: Master Machine Learning Fundamentals by University of Washington
- Specialization: Statistics with R by Duke University
- Specialization: Software Development in R by Johns Hopkins University
- Specialization: Genomic Data Science by Johns Hopkins University
Popular Courses Launched in 2020
- Google IT Automation with Python by Google
- AI for Medicine by deeplearning.ai
- Epidemiology in Public Health Practice by Johns Hopkins University
- AWS Fundamentals by Amazon Web Services
Trending Courses
- The Science of Well-Being by Yale University
- Google IT Support Professional by Google
- Python for Everybody by University of Michigan
- IBM Data Science Professional Certificate by IBM
- Business Foundations by University of Pennsylvania
- Introduction to Psychology by Yale University
- Excel Skills for Business by Macquarie University
- Psychological First Aid by Johns Hopkins University
- Graphic Design by Cal Arts
Books - Data Science
Our Books
- Practical Guide to Cluster Analysis in R by A. Kassambara (Datanovia)
- Practical Guide To Principal Component Methods in R by A. Kassambara (Datanovia)
- Machine Learning Essentials: Practical Guide in R by A. Kassambara (Datanovia)
- R Graphics Essentials for Great Data Visualization by A. Kassambara (Datanovia)
- GGPlot2 Essentials for Great Data Visualization in R by A. Kassambara (Datanovia)
- Network Analysis and Visualization in R by A. Kassambara (Datanovia)
- Practical Statistics in R for Comparing Groups: Numerical Variables by A. Kassambara (Datanovia)
- Inter-Rater Reliability Essentials: Practical Guide in R by A. Kassambara (Datanovia)
Others
- R for Data Science: Import, Tidy, Transform, Visualize, and Model Data by Hadley Wickham & Garrett Grolemund
- Hands-On Machine Learning with Scikit-Learn, Keras, and TensorFlow: Concepts, Tools, and Techniques to Build Intelligent Systems by Aurelien Géron
- Practical Statistics for Data Scientists: 50 Essential Concepts by Peter Bruce & Andrew Bruce
- Hands-On Programming with R: Write Your Own Functions And Simulations by Garrett Grolemund & Hadley Wickham
- An Introduction to Statistical Learning: with Applications in R by Gareth James et al.
- Deep Learning with R by François Chollet & J.J. Allaire
- Deep Learning with Python by François Chollet